Description
This is the Quick Start Guide on how to run the stable version of the MMB UMCU Tourmaline on the HPC, consisting of a quick overview of QIIME 2, a setup part and instructions of how to run the pipeline.
Overview of QIIME 2
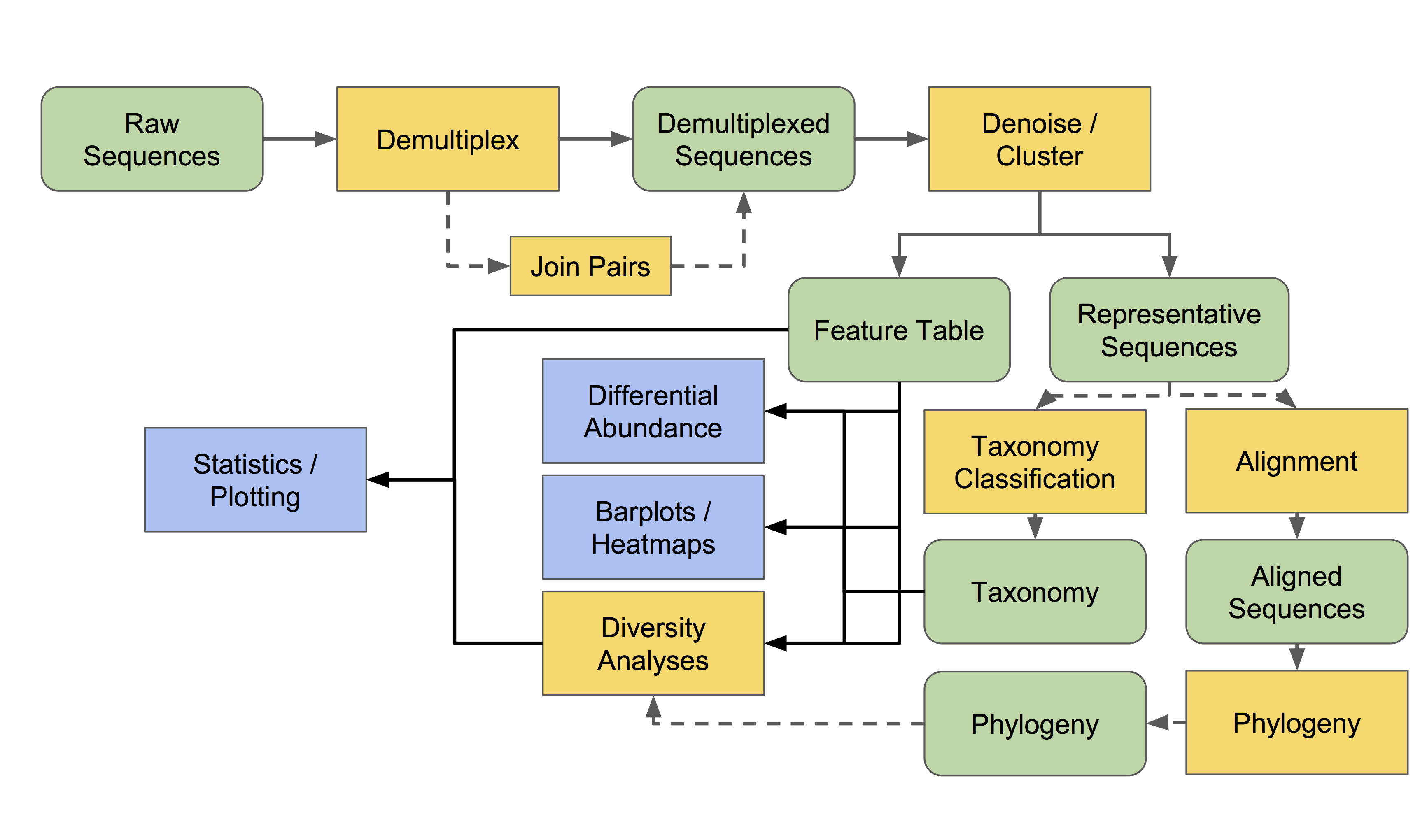
The core commands of Tourmaline are all commands of QIIME 2, one of the most popular amplicon sequence analysis software tools available. Below is a workflow diagram of QIIME 2. For more in depth information, visit this page.
Setup
Clone the Tourmaline repository into your data/ folder on the HPC and navigate to the cloned Tourmaline folder. For this purpose you need a gitlab account. Instructions to create an account, see http://143.121.18.159/doku.php?id=intro_to_git_gitlab
cd data/
git clone --depth 1 --branch v2.3 https://gitlab.com/malbert.rogers/tourmaline.git tourmaline_project_name
cd tourmaline_project_name/
Replace ‘tourmaline_project_name’ with the name of your project.
HAVING TROUBLE WITH GIT CLONE? In case git clone does not work, you could try to download the reporsitory as a zip-file using the Download button (next to the Clone button), transfer this file to the HPC and unzip.
Running the pipeline
There are some prerequisites that need to be followed before actually starting the pipeline.
Transfer files
Transfer the readfiles (R1.fastq.gz and R2.fastq.gz) and the metadata file (.txt)
to the cloned folder
tourmaline_project_name/ using WinSCP (for instuctions see Transfer
Files Using
WinSCP ) or FileZilla (for MacBook).
FastQC
After transfer of the readfiles to the HPC, it is recommended to first perform a quality control on the reads using FASTQC (for instructions see FastQC for read-quality control ), to assess the quality of the readfiles. This will be necessary to determine the truncation values in the config file of the tourmline pipeline.
Lay-out metadata and validation
There are a few prerequisites for the metadata file to avoid errors running the pipeline. In general, if your aim is to run Tourmaline to obtain the taxonomy and feature tables and perform the statistical analysis in R, the advise is to make a metadata file that contains only three columns: 1) header should be ‘sample_name’ 2) ‘BarcodeSequence’ and 3) ‘any name’, this header will be used for the beta_group_column (see below in ‘Edit config.yaml’). For column 3 you could use as header e.g. sample_source. The data in this column are not actually used. Note: for the Tourmaline pipeline the LinkerPrimerSequence is not necessary.
Important: don’t use special characters, only dots, underscores and dashes are allowed. Validate the metadata file and check for possible warnings/errors:
./validate_metadata.py metadata_file_example.txt
After validation a new metadata.tsv file has been generated in the tourmaline_project_name/00-data folder. Please check before starting the pipeline. Alternative: in case you are 100% sure that your metadata file doesn’t contain special characters, you can skip the validation step and directly transfer the file to the tourmaline_project_name/00-data folder. However, make sure that this file contains an .tsv extension. This folder always already contains a metadata file for the mock R1 and R2.fastq files in case you want to test the pipeline. However, you can overwrite this.
Edit config.yaml
Open and edit the configuration file, config.yaml:
nano config.yaml
Edit the the values for the following parameters:
R1_fastq_file: ## 1-mock_data_pool_R1.fastq.gz is filled in as default
R2_fastq_file: ## 1-mock_data_pool_R2.fastq.gz is filled in as default
dada2_trunc_len_f: ## Truncation value for the original R2 (reverse) readfile, default = 225
dada2_trunc_len_r: ## Truncation value for the original R1 (forward) readfile, default = 245
beta_group_column: ## Any header of the metadata file, default = Type
R1_fastq_file: forward (R1) readfile name including all extensions (example_L001_R1_001.fastq.gz)
R2_fastq_file: reverse (R2) readfile name including all extensions (example_L001_R2_001.fastq.gz)
dada2_trunc_len_f: truncation value for the R2 (reverse) readfile. This should be determined based on the results from a FastQC report. All bases after this value will be trimmed for all R2 reads. Please note that the reads in the FastQC report still contain barcode and primer sequences, while at this point in the Tourmaline pipeline both were already removed. Consequently, the reads would be approximately 36 bases (+/- 3) shorter from the front, resulting in a total length of around 264 bases.
dada2_trunc_len_r: truncation value for the R1 (forward) readfile. This should be determined based on the results from a FastQC report. All bases after this value will be trimmed for all R1 reads. Please note that the reads in the FastQC report still contain barcode and primer sequences, while at this point in the Tourmaline pipeline both were already removed. Consequently, the reads would be approximately 36 bases (+/- 3) shorter from the front, resulting in a total length of around 264 bases.
beta_group_column: a column header of the metadata file. This is required for the pipeline to run. You could pick a random column header or a column header of interest, if you would also like alpha- and beta-diversity analyses performed (on this specific column).
Optional: In the case that you would also like Tourmaline to generate alpha- and beta-diversity outputs, it’s then recommended to also adapt the values for following parameters (more information on these parameters can be found in the configuration file):
core_sampling_depth: ## Minimum number of sequences required for a sample to be included in further diversity analyses. Default = 500
alpha_max_depth: ## Maximum number of sequences for alpha diversity calculations. Also used as a maximum for the rarefaction curve. Default = 10000
Save and quit nano. You do this by pressing Ctrl + X (to quit), then press Y (to save) and finally press ENTER.
Run Tourmaline
Now the pipeline can be executed. For QIIME2 analyses to obtain features-tables and taxonomy* outputs, run the following command (see below for other possible options):
./sbatch_tourmaline.sh taxonomy
*Input options for running the pipeline are:
demux: only demultiplex the sequencing data, generating seperate fastq-files for each sample (recommended as a first step for projects’ data that are split over multiple sequencing runs);
denoise: imports FASTQ data and runs denoising, generating a feature table and representative sequences;
taxonomy: assigns taxonomy to representative sequences (recommended option to run in most cases);
diversity: step does representative sequence curation, core diversity analyses, and alpha and beta group significance;
report: step generates an HTML report of the outputs plus metadata, inputs, and parameters. Also, the report step can be run immediately to run the entire workflow.